Thalassemia is a genetic disease infecting four out of every 100,000 people in the United States alone. Read and know all about the disorder, including its possible types, causes, symptoms, diagnosis, treatment and more.
What is Thalassemia?
Page Contents
- 1 What is Thalassemia?
- 2 Thalassemia ICD9 Code
- 3 Thalassemia Types
- 4 Thalassemia Symptoms
- 5 Thalassemia Causes
- 6 Thalassemia Risk Factors
- 7 Thalassemia Diagnosis
- 8 Thalassemia Differential Diagnoses
- 9 Thalassemia Treatment
- 10 Thalassemia Prognosis
- 11 Thalassemia Complications
- 12 Thalassemia Prevention
- 13 Thalassemia Coping and Support
- 14 Thalassemia Pictures
It is an inherited blood disease in which the body produces an abnormal type of hemoglobin – the protein present in the red blood cells that transports oxygen. In patients of Thalassemia, there is excessive destruction of RBCs (red blood cells) which results in anemia.
The condition is referred to by various other names, such as:
- Cooley’s anemia
- Alpha Thalassemia
- Beta Thalassemia
- Mediterranean anemia
Thalassemia ICD9 Code
The ICD9 Code for this disorder is 282.4.
Thalassemia Types
The condition is mainly differentiated into the following two types:
Alpha Thalassemia
It arises when one or more genes associated to the alpha globin protein are either changed or missing. It most commonly occurs in individuals from China, Middle East, southeast Asia and Africa.
Beta Thalassemia
It develops when similar genetic impairments affect the manufacture of the beta globin protein. It occurs in people of Mediterranean descent and to a lesser extent, Asians, Chinese and African Americans.
Both Alpha and Beta forms of Thalassemia can be classified into the following two subtypes:
Thalassemia Major
This type develops when an individual inherits the impaired gene from both parents. Beta Thalassemia Major is also known as Cooley’s anemia.
Thalassemia Minor
It arises if a person gets the impaired gene from only one parent. People with this type of the disease are carriers of the condition and generally do not experience any signs and symptoms.
Thalassemia Symptoms
The signs and symptoms of this disorder may include the following problems:
- Irritability
- Dark urine
- Fatigue
- Weakness
- Slow growth
- Abdominal swelling
- Pale appearance
- Shortness of breath
- Facial bone deformities
- Yellow discoloration of skin (indicative of Jaundice)
The symptoms experienced by sufferers are based on the form and severity of the condition. Some babies may manifest the symptoms at birth while others may develop its signs during the first two years after birth. Certain individuals suffering from only one affected hemoglobin gene do not experience any symptoms of Thalassemia.
Alpha Thalassemia Symptoms
Four genes are associated in the production of the alpha hemoglobin chain. Patients receive two genes from each parent. The signs and symptoms may differ on the basis of the number of genes acquired by patients:
- Patients do not manifest any signs of Thalassemia in case of only one mutated gene. However, a carrier of the disorder can pass the gene on to children.
- The signs are mild in case of two mutated genes. This form of the disorder may be referred to as Alpha- Thalassemia minor. Patients may also be said to show an Alpha- Thalassemia trait.
- The symptoms may range from moderate to severe in case of three mutated genes. This type of the disorder is also known as Hemoglobin H disease.
- In case of four mutated genes, the disease is referred to as Hydrops fetalis or Alpha-Thalassemia Major. It generally leads to the death of a fetus prior to delivery or right after birth.
Beta Thalassemia Symptoms
Two genes are associated in the manufacture of the beta hemoglobin chain. A person may acquire one gene from each parent. The symptoms may depend on the number of genes received by sufferers:
In case of one mutated gene, sufferers may have mild symptoms. This type of the disease is known as Beta- Thalassemia minor or called a Beta-Thalassemia trait.
Acquiring two mutated genes may lead patients to suffer from symptoms that range from moderate to severe in nature. The form of the disorder is referred to as Cooley’s anemia or Beta-Thalassemia major. Babies who are born with two impaired beta hemoglobin genes are generally healthy at birth. However, they develop the signs within the first two years after birth.
Thalassemia Causes
The disorder arises due to mutations in the DNA of those cells that manufacture hemoglobin, the protein transporting oxygen in the body. The mutations that are related with Thalassemia are passed on to children from parents.
The mutations resulting in Thalassemia disrupt the normal production of hemoglobin and lead to low levels of the protein. As a consequence, this results in a high rate of destruction of red blood cells and leads to anemia. In anemic individuals, there are not enough amounts of red blood cells (RBCs) to transport oxygen to the tissues. This leaves sufferers fatigiued.
Thalassemia Risk Factors
The factors that increase the risk of suffering from this condition include:
Being of certain origin
The condition most often arises in individuals of Greek, Asian, African, Middle Eastern and Italian ancestry.
Having a family history
The disease is passed on to children from parents through mutated hemoglobin genes. Having a family history of the condition can elevate the risk of the disorder in individuals.
Thalassemia Diagnosis
The majority of children exhibiting moderate to severe form of Thalassemia exhibit the symptoms within the initial two years after birth. The diagnostic tests for this disorder may include:
Physical examination
A physical examination may reveal an enlargement of the spleen.
Blood tests
If a physician suspects the presence of Thalassemia, he or she may recommend blood tests to confirm the diagnosis.
In case of the presence of Thalassemia, blood tests may show:
- Paleness of RBCs (Red blood cells)
- Low level of RBCs
- Smaller appearance and abnormal shape of red blood cells under microscopic examination
- Uneven distribution of hemoglobin in red blood cells, which gives a bull’s eye appearance to the cells under the microscope
Blood tests may also be used for the following purposes:
- Evaluation of the hemoglobin
- Measurement of the amount of iron in blood
- Performing DNA analysis to detect Thalassemia or to assess whether or not an individual is carrying mutated hemoglobin genes
Prenatal testing
Before the birth of a baby, this type of testing can be done to determine the presence of Thalassemia and how severe it is. Tests used to detect the condition in fetuses include:
Amniocentesis
The test is generally conducted around the 11th week of pregnancy and involves the removal of a small piece of placenta for assessment.
Chorionic villus sampling
The test is generally carried out around the 16th week of pregnancy. It involves extracting a sample of the fluid surrounding the fetus.
Assisted reproductive technology
This procedure combines pre-implantation genetic diagnosis along with in-vitro fertilization and may assist parents having Thalassemia or the carriers of an impaired hemoglobin gene who give birth to healthy infants. The method includes retrieving mature eggs from a woman and fertilizing them in a laboratory dish with a male sperm. The embryos are tested to determine the presence of impaired genes. Only those not having genetic impairments are implanted in the woman.
Thalassemia Differential Diagnoses
The differential diagnoses of this disorder include distinguishing its signs from those of disorders that give rise to similar symptoms:
- Leukemia
- Iron deficiency anemia
- Hemoglobinopathy
- Other types of Thalassemia
Doctors must ensure that the signs of the above disorders are not what patients suspected of Thalassemia are actually complaining about.
Thalassemia Treatment
The treatment for the condition depends on the type of the disorder that a patient suffers from as well as its intensity. In mild Thalassemia, symptoms are usually mild and little or no treatment is required. A blood transfusion may be needed in the following cases:
- After an operation
- After giving birth to a baby
- To help control the symptoms of the disease
The treatment for moderate to severe cases of the disease may involve:
Stem cell transplant
The process is also referred to as “Bone Marrow Transplant.” In selected cases, it may be used to cure severe Thalassemia. Before this procedure, extremely high doses of radiation or drugs may be needed to destroy the diseased bone marrow in sufferers. Following this, patients may receive stem cell infusions from a compatible donor. However, this method can involve acute risks for sufferers which can also include death. Due to this reason, Stem cell transplant is generally reserved for patients with the most acute form of the disorder having a compatible donor – generally a sibling.
Frequent blood transfusions
Frequent blood transfusions are often needed for more acute forms of Thalassemia, possibly in every few weeks. However, this may cause buildup of excess iron in the body and medicines may be needed to get rid of it.
Thalassemia Prognosis
Acute cases of this disorder can lead to early death of sufferers as a result of cardiac failure, generally between 20 and 30 years of age. Regular blood transfusions and therapy can help remove excess iron from the body and improve the outcome of the disease.
The lifespan is generally normal in less severe types of the disease.
In people with a family history of the disorder who are planning to have children, prenatal screening and genetic counseling can be helpful.
Thalassemia Complications
If left untreated, the condition can result in cardiac failure and liver problems. It can also make patients susceptible to infections. Some of the symptoms can be managed with blood transfusions although they may lead to high levels of iron in the body which can cause damage to the endocrine system, liver and heart.
Thalassemia Prevention
The disorder cannot be prevented in the majority of cases. In people having Thalassemia or carrying a gene responsible for the disease, consultation with a genetic counselor can provide patients with valuable guidance before having a child.
Thalassemia Coping and Support
Coping with this disorder can be quite challenging. However, patients can find useful information, guidance and assistance with the various support groups and forums existing online and off the web. You can consult your doctor to know about support groups in your area or get in touch with the Cooley’s Anemia Foundation at 800-522-7222.
Thalassemia Pictures
The images below can help you get a visual idea about the disorder.
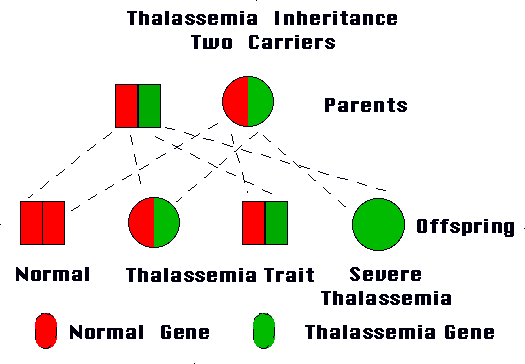
Picture 1 – Thalassemia
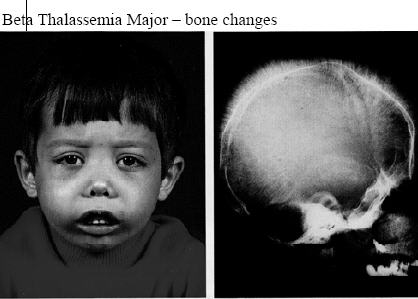
Picture 2 – Thalassemia Image
If you suspect yourself or your child to be having the symptoms of Thalassemia, get in touch with a professional medical care provider as soon as possible. Left untreated, the disorder can wreak havoc on your system and leave you susceptible to various problems some of which can even be life-threatening. Naturally, it is best to seek early diagnosis and treatment which can ensure better management of the problems.
References:
http://www.mayoclinic.com/health/thalassemia/DS00905
http://www.nlm.nih.gov/medlineplus/ency/article/000587.htm
http://www.umm.edu/ency/article/000587.htm
http://www.mdguidelines.com/thalassemia
http://en.wikipedia.org/wiki/Thalassemia