Craniodiaphyseal dysplasia Definition
Page Contents
- 1 Craniodiaphyseal dysplasia Definition
- 2 Craniodiaphyseal dysplasia History
- 3 Craniodiaphyseal dysplasia Symptoms
- 4 Craniodiaphyseal dysplasia Causes
- 5 Craniodiaphyseal dysplasia Diagnosis
- 6 Craniodiaphyseal dysplasia Treatment
- 7 Craniodiaphyseal dysplasia Management and Control
- 8 Craniodiaphyseal dysplasia Prognosis
- 9 Craniodiaphyseal dysplasia Complications
- 10 Craniodiaphyseal Dysplasia Support Group
- 11 Craniodiaphyseal dysplasia Pictures
- 12 Craniodiaphyseal dysplasia ICD10 Code
Craniodiaphyseal dysplasia is a very rare genetic disorder of the bone [1]. Only around 30 cases of its have been reported so far. It is also known as lionitis or simply as CDD.
Craniodiaphyseal dysplasia History
Cases with features similar to CDD were first reported in 1949 (by Halliday) and in 1958 (by Joseph et al). It was then regarded as a type of Camurati-Engelmann disease. It was not until in 1969 that it was first described by Gorlin et al as a very acute bone disease characterized by massive hyperostosis and sclerosis, particularly affecting the facial and skull bones.
CDD became known through the 1985 drama film “Mask” by Peter Bogdanovich that showed the case of an American boy Roy L. Dennis aka Rocky who died of the disease in 1978.
Craniodiaphyseal dysplasia Symptoms
There is buildup of calcium within the skull, which reduce the size of cranial foramina. This leads to abnormal facial features and lowers the lifespan of patients. The calcium deposits can also reduce the hole in the cervical spinal canal. There is compression of cranial nerves, the intracranial contents and the foramen magnum, which commonly results in:
- Blindness
- Epilepsy
- Facial diplegia
- Loss of hearing
- Mental retardation
Stenosis of the cervical canal may cause quadriparesis (a late complication of hyperostosis). Sclerosis of the expanded diaphyses and hyperostosis can be observed in the long bones. In some cases, a postural defect in the metaphyses may also be noted.
Craniodiaphyseal dysplasia Causes
It is supposed to be hereditary, although the underlying genetics is unclear. The disease is acquired in an autosomal recessive fashion. Very few cases of CDD have been described, which causes difficulty in determining genetic association.
Craniodiaphyseal dysplasia Diagnosis
A difference in the size of eyes is a common reported sign for CDD. Physicians also look for clinical symptoms like:
- Dacryocystitis [2]
- Seizures
- Mental retardation
- Paralysis
These are complications ensuing as a result of diminutive foramina. X-ray images can show regions of abnormal bone density, where excess calcium deposition occurs and many osteoblasts and cells that manufacture new bones are present.
Craniodiaphyseal dysplasia Differential Diagnosis
Mutation analysis of the gene TGFB1 lets doctors rule out the presence of Camurati-Engelmann disease, the signs of which are similar to those of CDD. Other disorders to be considered include:
- Craniotubular dysplasias
- Van Buchem’s Dysplasia
Craniodiaphyseal dysplasia Treatment
CDD sufferers, particularly those with tracheal intubation problems and difficulties with airway management, need to be taken to an anesthetist. Decompressing craniectomy can be used to enlarge the middle and the anterior fossae in case of acute brain compression with signs of elevated intracranial pressure.
In case of papilledema, decompression of optic and orbital nerve may be needed for cure. Calcitriol/calcitonin therapy, along with prednisone or low calcium diet may change the clinical course of progression [3].
Long-term treatment is based on controlling rapid malformation of bones.
Craniodiaphyseal dysplasia Management and Control
Further progression of CDD can be managed by close observation of the health of sufferers. Magnetic Resonance Imaging (MRI) should be used as a routine component for evaluating patients.
Craniodiaphyseal dysplasia Prognosis
The outcome is poor. Generally, benefits from dacryocystorhinostomy, craniofacial remodeling and choanal stenosis surgery persist only for a short time. Reconstitution of osseous optic canals occurs rather rapidly, and there may be a recurrence of papilledema. Most patients have been found to die in childhood in the few reported cases of CDD. Life expectancy is extremely bad for patients with this disease. It is vital to recognize these conditions on an early basis as therapy directed at the underlying bony defect can succeed the best if started during infancy.
Craniodiaphyseal dysplasia Complications
The probable complications include:
- Affected vascular supply (due to hyperostosis)
- Severe facial deformity
- Elevated numbers of osteoblasts
- Loss of vision [4]
- Deformation of ossicles due to bony overgrowth
- Significant brain compression
- Epilepsy
- Complete hearing inability
- Reduced air spaces of mastoid, middle ear cavity and external auditory canal by hyperostotic bone
- Facial diplegia
- Dysfunction of facial nerves
- Problems in air conduction, due to anomalous ossicles with hyperostosis
- Moderate sclerosis and postural defects (in clavicles, ribs and pelvis)
- Retarded mental abilities
- Quadriparesis
- Mechanical damage of nerve fibers
- Nasolacrimal obstruction
Craniodiaphyseal Dysplasia Support Group
For information and guidance, family members of CDD sufferers may contact:
Office of Rare Diseases Research
National Center for Advancing Translational Sciences (NCATS)
National Institutes of Health
6701 Democracy Boulevard
Suite 1001, MSC 4874
Bethesda, MD 20892
For courier use (Bethesda, MD 20817)
(301) 402-4336 (voice)
(301) 480-9655 (fax)
Email: [email protected]
Craniodiaphyseal dysplasia Pictures
Check these photos and know about the physical appearance of patients with CDD from the images.
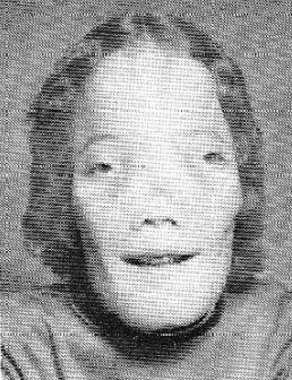
Picture 1 – Craniodiaphyseal dysplasia

Picture 2 – Craniodiaphyseal dysplasia Image
Craniodiaphyseal dysplasia ICD10 Code
The ICD10 Code for this disorder is M85.2.
References:
Craniodiaphyseal dysplasia (Wikipedia)
Craniodiaphyseal dysplasia (Piczo.com)
Craniodiaphyseal dysplasia (Orpha.net)
What is Craniodiaphyseal dysplasia? (Wisegeek)